


Next: Summary Up: Results Previous: A Dijkstra-based Selector Contents
Tryptophan zippers are stable fast-folding 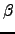 -hairpins designed by Cochran et al. [156], which have recently generated considerable interest [157,158]. In the present work we have obtained native to denatured state rearrangement pathways for five tryptophan zippers: trpzip
-hairpins designed by Cochran et al. [156], which have recently generated considerable interest [157,158]. In the present work we have obtained native to denatured state rearrangement pathways for five tryptophan zippers: trpzip 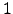 , trpzip
, trpzip 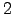 , trpzip
, trpzip 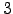 , trpzip -I and trpzip
, trpzip -I and trpzip 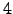 . The notation is adopted from the work of Du et al. [158]. All these peptides contain twelve residues, except for trpzip , which has sixteen. Tryptophan zippers , , and -I differ only in the sequence of the turn. Experimental measurements of characteristic folding times for these peptides have shed some light on the significance of the turn sequence in determining the stability and folding kinetics of peptides with the -hairpin structural motif [158].
. The notation is adopted from the work of Du et al. [158]. All these peptides contain twelve residues, except for trpzip , which has sixteen. Tryptophan zippers , , and -I differ only in the sequence of the turn. Experimental measurements of characteristic folding times for these peptides have shed some light on the significance of the turn sequence in determining the stability and folding kinetics of peptides with the -hairpin structural motif [158].
To model these molecules we used a modified CHARMM19 force field [4], with symmetrised Asn, Gln and Tyr dihedral angle and Cter improper dihedral angle terms, to ensure that rotamers of these residues have the same energies and geometries. These changes to the standard CHARMM19 force field are described in detail in Appendix A. Another small modification concerned the addition of a non-standard amino acid, D-proline, which was needed to model trpzip . The implicit solvent model EEF1 was used to account for solvation [11], with a small change to the original implementation to eliminate discontinuities [159].
We have used the Dijkstra-based connection algorithm to obtain folding pathways for all five trpzip peptides. In each case the first endpoint was chosen to be the native state structure, which, for , and trpzips, was taken from the Protein Data Bank (PDB) [160]. There are no NMR structures available for and -I, so for these peptides the first endpoint was chosen to be the putative global minimum obtained using the basin-hopping method [161,162,163]. The folded state for trpzip is depicted in Figure 2.10. The total charge of this molecule is 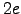 (two Lys)
(two Lys)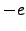 (Glu)
(Glu)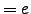 . However, in our calculations the total charge was zero because in the CHARMM19 force field ionic sidechains and termini are neutral when the EEF1 solvation model is used [11].
. However, in our calculations the total charge was zero because in the CHARMM19 force field ionic sidechains and termini are neutral when the EEF1 solvation model is used [11].
Figure: The folded state of tryptophan zipper 2 (shown in stereo). This structure contains 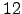 residues and two terminal capping groups, containing a total of
residues and two terminal capping groups, containing a total of 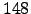 CHARMM19 atoms. The sequence of residues is Ace-Ser-Trp-Thr-Trp-Glu-Asn-Gly-Lys-Trp-Thr-Trp-Lys-Cbx. The naming convention for amino acid residues is the same as in Reference BrandenT99. N- and C-termini are capped with the standard CHARMM19 blocking groups, `Ace' (-CO-CH
CHARMM19 atoms. The sequence of residues is Ace-Ser-Trp-Thr-Trp-Glu-Asn-Gly-Lys-Trp-Thr-Trp-Lys-Cbx. The naming convention for amino acid residues is the same as in Reference BrandenT99. N- and C-termini are capped with the standard CHARMM19 blocking groups, `Ace' (-CO-CH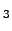 ) and `Cbx' (-NH-CH), respectively. The structure was obtained by minimising the first out of 20 structures deposited in the Protein Data Bank (PDB ID: 1LE1) by Cochran, Skelton, and Starovasnik [156]. The RMS force and modified [165] CHARMM19 energy are
) and `Cbx' (-NH-CH), respectively. The structure was obtained by minimising the first out of 20 structures deposited in the Protein Data Bank (PDB ID: 1LE1) by Cochran, Skelton, and Starovasnik [156]. The RMS force and modified [165] CHARMM19 energy are 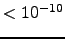 kcalmol
kcalmol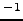 Å and
Å and 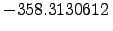 kcalmol, respectively. The structural motif of this de novo peptide is a -hairpin. It can be seen that the backbone is stabilised by a U-turn and six hydrogen bonds while four hydrophobic tryptophan sidechains are packed nicely on the side. Red, blue and light grey balls denote oxygen, nitrogen and hydrogen atoms, respectively. Dark grey balls denote carbon atoms as well as CHARMM19 united atoms of types CH, CH2 and CH3. Ace and Cbx each contain a single united atom of type CH3. There is one CH2 `atom' in each of the four tryptophans (connecting the 5-membered ring of the sidechain with the backbone). This figure was prepared using MolMol [166].
kcalmol, respectively. The structural motif of this de novo peptide is a -hairpin. It can be seen that the backbone is stabilised by a U-turn and six hydrogen bonds while four hydrophobic tryptophan sidechains are packed nicely on the side. Red, blue and light grey balls denote oxygen, nitrogen and hydrogen atoms, respectively. Dark grey balls denote carbon atoms as well as CHARMM19 united atoms of types CH, CH2 and CH3. Ace and Cbx each contain a single united atom of type CH3. There is one CH2 `atom' in each of the four tryptophans (connecting the 5-membered ring of the sidechain with the backbone). This figure was prepared using MolMol [166]. 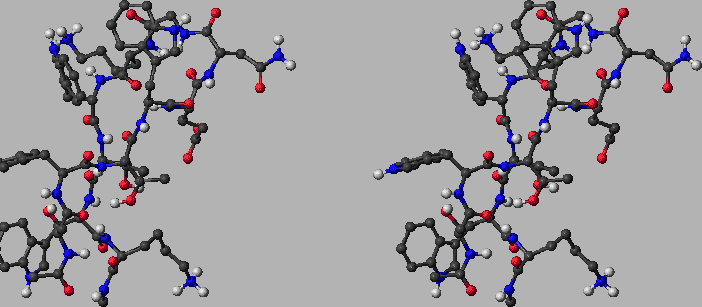 |
The second endpoint was chosen to be an extended structure, which was obtained by simply minimising the energy of a conformation with all the backbone dihedral angles set to 180 degrees. All the stationary points (including these obtained during the connection procedure) were tightly converged to reduce the root-mean-squared force below 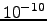 kcalmolÅ. The unfolded state for trpzip is depicted in Figure 2.11 for example.
kcalmolÅ. The unfolded state for trpzip is depicted in Figure 2.11 for example. Figure: The unfolded state of tryptophan zipper 2 (shown in stereo). This structure was obtained by minimising a conformation with all the backbone dihedral angles set to 180 degrees. The energy of this structure is 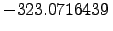 kcalmol. The RMS deviation from the minimised PDB structure depicted in Figure 2.10 is
kcalmol. The RMS deviation from the minimised PDB structure depicted in Figure 2.10 is 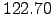 Å. See the caption of Figure 2.10 for notation and other details.
Å. See the caption of Figure 2.10 for notation and other details. 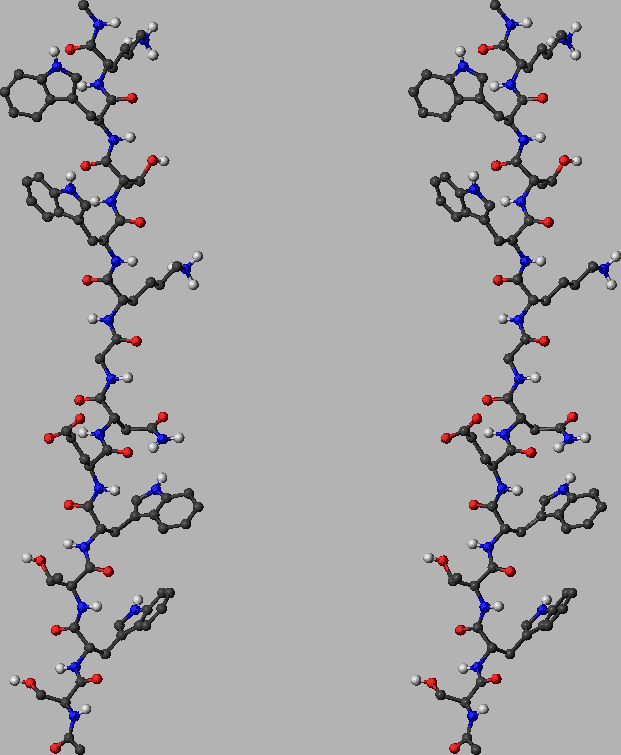 |
Each of the five trpzip pathway searches was conducted on a single Pentium 4 3.0GHz 512Mb cache CPU and required less than 24 hours of CPU time. The timings could certainly be improved by optimising the various parameters employed throughout the searches. However, it is more important that the connections actually succeed in a reasonably short time. It only requires one complete path to seed a DPS run, and we expect the DPS procedure to reduce the length of the initial path by a least a factor of two in sampling the largest contributions to the effective two-state rate constants. The results of all the trpzip calculations are shown in Figure 2.12.
| Trpzip | Steps | Cycles | Searches | Points | | 1 | 70 | 44 | 190 | 161,137 | | 2 | 93 | 386 | 1325 | 761,626 | | 3 | 128 | 203 | 703 | 565,487 | | 3-I | 67 | 52 | 172 | 203,158 | | 4 | 91 | 135 | 558 | 284,244 | |
Figure: Energy profiles for native to denatured state rearrangements of tryptophan zippers found by the Dijkstra-based connection algorithm. For each profile the number of steps in the pathway, the number of connection algorithm cycles, the total number of DNEB searches and the total number of stationary points in the database (recorded upon termination of the algorithm) are shown. The total number of stationary points is presented in the form 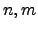 , where
, where 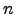 is the number of minima and
is the number of minima and 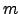 is the number of transition states. The potential energy,
is the number of transition states. The potential energy, 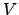 , is given in the units of kcal/mol, and the integrated path length,
, is given in the units of kcal/mol, and the integrated path length, 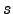 , is given in the units of Å.
, is given in the units of Å.
Next: Summary Up: Results Previous: A Dijkstra-based Selector Contents Semen A Trygubenko 2006-04-10 ![\begin{psfrags}\psfrag{s} [bc][bc]{$s$}
\psfrag{V} [bc][bc]{$V$}
\psfrag{t...
...}
\includegraphics[height=0.25\textheight]{dneb/trpzipEofS/1.eps}
\end{psfrags}](img503.png)
![\begin{psfrags}
\psfrag{s} [bc][bc]{$s$}
\psfrag{V} [bc][bc]{$V$}
\psfrag{...
...
\includegraphics[height=0.25\textheight]{dneb/trpzipEofS/2.eps}
\end{psfrags}](img504.png)
![\begin{psfrags}
\psfrag{s} [bc][bc]{$s$}
\psfrag{V} [bc][bc]{$V$}
\psfrag{...
...
\includegraphics[height=0.25\textheight]{dneb/trpzipEofS/3.eps}
\end{psfrags}](img505.png)
![\begin{psfrags}
\psfrag{s} [bc][bc]{$s$}
\psfrag{V} [bc][bc]{$V$}
\psfrag{...
...\includegraphics[height=0.25\textheight]{dneb/trpzipEofS/3-I.eps}
\end{psfrags}](img506.png)
![\begin{psfrags}
\psfrag{s} [bc][bc]{$s$}
\psfrag{V} [bc][bc]{$V$}
\psfrag{...
...
\includegraphics[height=0.25\textheight]{dneb/trpzipEofS/4.eps}
\end{psfrags}](img507.png)